By Tidyomics team | December 9, 2018
We need to be aware that there are two genomics coordinate systems: 1 based and 0 based. There is really no mystery between these two. You EITHER count start at 0 OR at 1. However, this can make confusions when analyzing genomic data and one may make mistakes if not keep it in mind.
The TWO systems
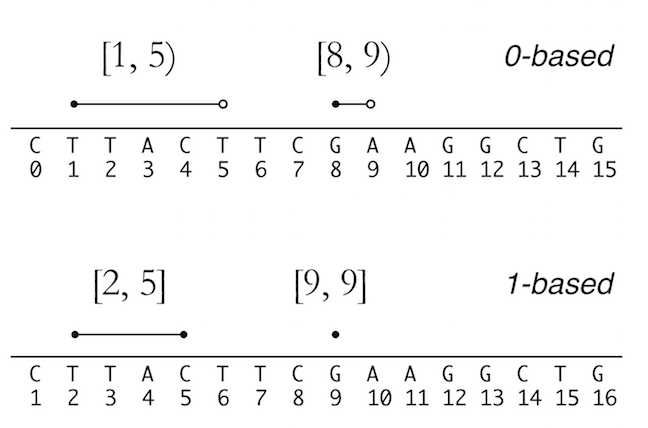
See the figure below to understand the two systems. credit due to Vince Buffalo
from his book Bioinformatics data skills.
There is a nice digital cheat sheet from this post by Obi Griffith, a professor of Medicine (Oncology) and Genetics at Washington University five years ago. We frequently go back to reference it.

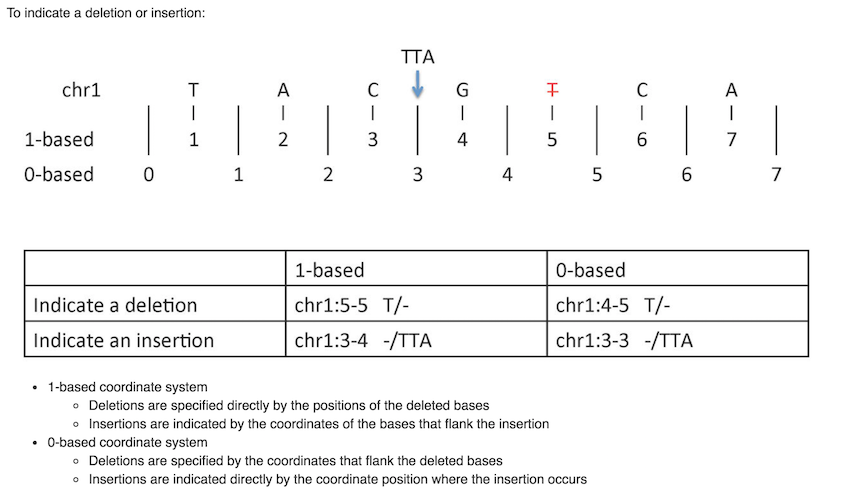
Mistakes can happen if not consider the coordinate system carefully
The problem is that different file format uses different coordinate systems. Well, you know it is bioinFORMATics :)
some files such as bed file
is 0 based. Two genomic regions:
chr1 0 1000
chr1 1000 2000
when you import that bed file into R using rtracklayer::import(), it will become
chr1 1 1000
chr1 1001 2000
The function converts it to 1 based internally (R is 1 based unlike python).
When you read the bed file with read.table and use GenomicRanges::makeGRangesFromDataFrame() to convert it to a GRanges object,
do not forget to add 1 to the start before doing it.
Similarly, when you write a GRanges object to disk using rtracklayer::export, you do not need to worry, R will convert it back to 0 based in file.However, if you make a dataframe out of the GRanges object, and write that dataframe to file, remember to do start -1 before doing it.
A full list of coordinate systems for different files can be found below from Bioinformatics data skills.
| Format/library | Type |
|---|---|
| BED | 0-based |
| GTF | 1-based |
| GFF | 1-based |
| SAM | 1-based |
| BAM | 0-based |
| VCF | 1-based |
| BCF | 0-based |
| Wiggle | 1-based |
| GenomicRanges | 1-based |
| BLAST | 1-based |
| GenBank/EMBL Feature Table | 1-based |
A note for UCSC genome browser
To make things worse, see FAQ from UCSC:
I am confused about the start coordinates for items in the
refGenetable. It looks like you need to add “1” to the starting point in order to get the same start coordinate as is shown by the Genome Browser. Why is this the case?Our internal database representations of coordinates always have a zero-based start and a one-based end. We add 1 to the start before displaying coordinates in the Genome Browser. Therefore, they appear as one-based start, one-based end in the graphical display. The
refGene.txtfile is a database file, and consequently is based on the internal representation.We use this particular internal representation because it simplifies coordinate arithmetic, i.e. it eliminates the need to add or subtract 1 at every step. If you use a database dump file but would prefer to see the one-based start coordinates, you will always need to add 1 to each start coordinate.
If you submit data to the browser in position format
chr#:##-##, the browser assumes this information is 1-based. If you submit data in any other format (BEDchr# ## ##) or otherwise), the browser will assume it is 0-based. You can see this both in our liftOver utility and in our search bar, by entering the same numbers in position or BED format and observing the results. Similarly, any data returned by the browser in position format is 1-based, while data returned in BED format is 0-based.For a detailed explanation, please see our blog entry for the UCSC Genome Browser coordinate counting systems.